










Neorabio量子化学服务基于先进的量子力学理论和计算方法,能够精确描述分子的电子结构、反应机理及能量变化。我们通过量子化学计算,帮助客户深入揭示分子间的相互作用和反应路径,准确评估结合能、反应势垒及分子性质,从而支持药物设计、材料科学及催化剂开发等领域。凭借Neorabio高性能计算资源和专业团队,我们提供包括能量优化、电子密度分析、激发态计算等多样化服务,助力客户提升分子设计的科学性和创新性,加快研发进程。

服务流程
[提交结构] → [结构预处理] → [参数设置] → [对接运行] → [结果分析] → [报告交付]
应用场景
用于药物分子设计中的结合能精确计算
解析小分子与靶点的电子结构及反应机理
辅助催化剂设计与反应路径优化
材料科学中新材料的电子性质预测
支持分子激发态及光物理性质的计算
指导分子改造和性质调控,提高活性和选择性
为多学科交叉研究提供高精度计算支持
技术优势
拥有先进的量子力学计算平台,支持多种高精度计算方法
具备完善的分子结构预处理和优化流程,确保计算的准确性和稳定性
支持多种复杂分子体系的模拟,包括大分子、多态结构及过渡态分析
结合高性能计算资源,能够快速处理大规模计算任务,提高效率
专业团队拥有丰富的计算化学与相关领域背景,提供深度分析与结果解读
提供灵活定制的服务方案,满足药物设计、材料科学及催化剂开发等多领域需求
注重计算结果与实验数据的结合,提升预测的可靠性和实用性
经典案例
静电势分析
分子轨道分析
光谱计算
势能面扫描
热力学性质计算
构象分析
分子芳香性和离域性分析
机反应机理计算
分子静电势(Electrostatic potential, ESP)分析通过可视化分子表面静电分布及其局部极值点,帮助研究人员理解分子的反应性及其与其他分子的相互作用,广泛应用于药物设计、催化剂开发和分子识别研究,能够帮助定位潜在的反应位点,研究分子与受体的相互作用模式,并为先导化合物的优化提供理论支持。
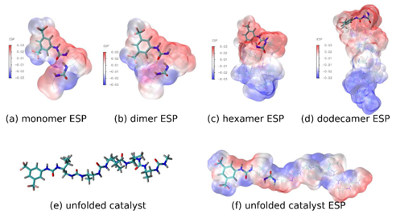
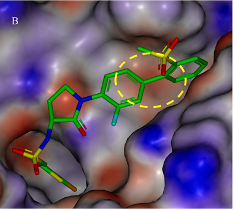
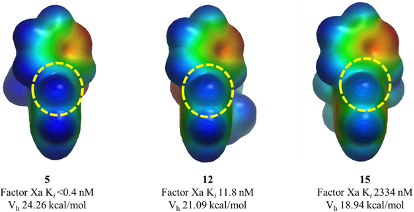
对催化剂的静电势表面分析【1】 对蛋白口袋的静电分布 不同化合物的静电分布强度及其活性【2】
【1】Insight into Substrate Recognition by Urea-Based Helical Foldamer Catalysts Using a DFT Global Optimization Approach. J. Org. Chem. 2022, 87 (16), 10726–10735. https://doi.org/10.1021/acs.joc.2c00562.
【2】Electrostatic Complementarity in Structure-Based Drug Design. J. Med. Chem. 2022, 65 (11), 7476–7488. https://doi.org/10.1021/acs.jmedchem.2c00164.
分子轨道分析基于分子轨道理论,深入探讨分子的电子结构,尤其是最高占据轨道(Highest Occupied Molecular Orbital, HOMO)和最低未占轨道(Lowest Unoccupied Molecular Orbital, LUMO)的特性。NoeraBio的分析能够帮助客户理解分子的光学和电子性质,预测化合物的反应活性和稳定性。
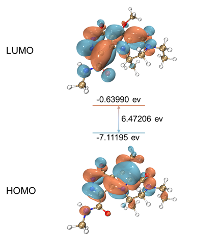
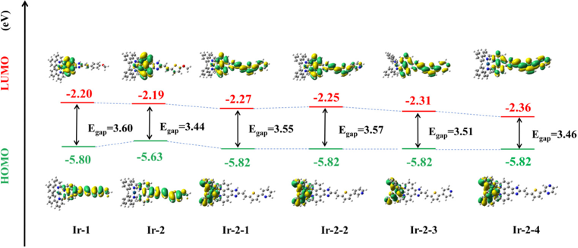
HOMO-LUMO gap计算 HOMO/LUMO等值面分析及HOMO-LUMO gap计算【3】
Theoretical Investigation of Novel Nitrogen-Heterocyclic Iridium(III) Polypyridyl Complexes as Photosensitizers for Two-Photon Photodynamic Therapy. J. Med. Chem. 2024, 67 (20), 18157–18169. https://doi.org/10.1021/acs.jmedchem.4c01292.
分子光谱计算利用量子化学方法,计算分子的红外(IR)、紫外可见光(UV-Vis)、圆二色谱(CD)等光谱特性。NeoraBio可以通过精确计算,预测分子的光谱峰位置和强度,帮助客户在实验前获得理论的光谱信息。
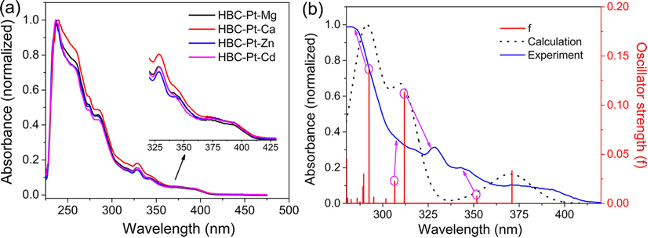
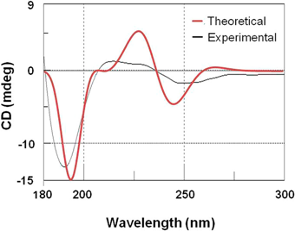
计算光谱得到跃迁的理论来源 【1】 计算电子圆二色谱辅助判断化合物绝对构型【2】
- Formation of Hetero-Binuclear Pt(II)-M(II) Complexes Based on (2-(1H-Tetrazol-5-Yl)Phenyl)Diphenylphosphine Oxide for Superior Phosphorescence of Monomers. Inorg. Chem. 2019, 58 (7), 4253–4261. https://doi.org/10.1021/acs.inorgchem.8b03326.
- Hippolachnin A, a New Antifungal Polyketide from the South China Sea Sponge Hippospongia Lachne. Org. Lett. 2013, 15 (14), 3526–3529. https://doi.org/10.1021/ol400933x.
分子势能面扫描服务通过绘制分子在不同构象或反应路径下的势能变化图,帮助研究人员理解分子结构变化所带来的能量变化。
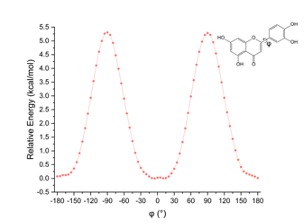
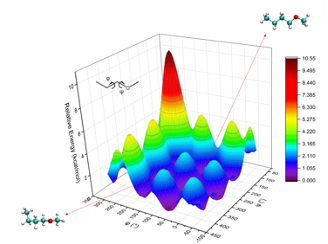
2D势能面扫描 3D势能面扫描 氯二氟乙酰氮杂烯(F2ClCC(O)N3)关键二面角的势能面扫描【1】
- Theoretical Insights into Photo-Induced Curtius Rearrangement of Chlorodifluoroacetyl Azide. Org. Chem. Front. 2017, 4 (6), 1153–1161. https://doi.org/10.1039/C7QO00083A.
分子热力学性质服务通过计算分子的热容、熵、焓和自由能等关键热力学参数,帮助研究人员全面了解分子的物理化学性质。NeoraBio的分析为客户提供了有关反应自发性、能量变化等关键特性的预测,优化反应条件。
pKa计算预测值与实验值比较
|
|
pyridine |
quinoline |
isoquinoline |
Pyrimidine |
2-Pyridinamine |
4-Pyridinamine |
3-Fluoropyridine |
|
Predicted pKa |
5.15 |
5.04 |
5.45 |
1.26 |
6.95 |
9.53 |
2.31 |
|
Experimental pKa |
5.23 |
4.93 |
5.42 |
1.3 |
6.9 |
9.3 |
2.97 |
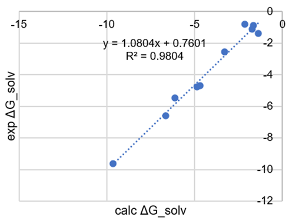
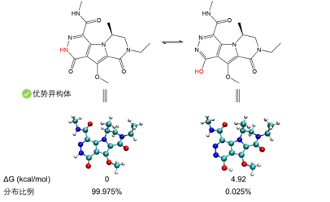

溶解自由能计算 异构体分析 计算确定Felbamate的最稳定异构体和构象【1】
- Tautomerism of the Antiepileptic Drug Felbamate: A DFT Study. J. Struct. Chem. 2017, 58 (2), 244–251. https://doi.org/10.1134/S0022476617020044.
分子构象分析旨在探索分子在不同条件下可能构象变化及其稳定性。此服务广泛应用于药物设计、分子对接、蛋白质-配体识别等领域,能够为优化分子构象、提升生物活性或化学稳定性提供理论支持,是新分子设计的重要依据。
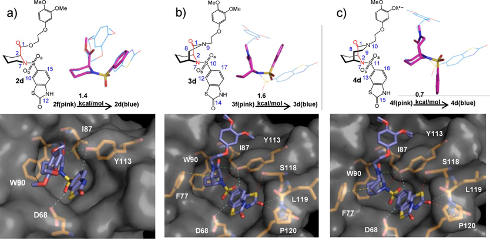
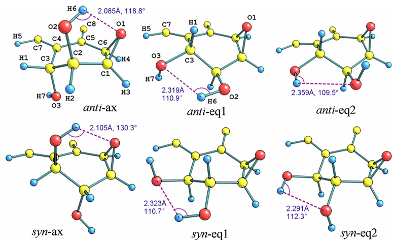
药物分子结合构象与低能构象的比较【1】 化合物最优构象的确证【2】
- Increasing the Efficiency of Ligands for FK506-Binding Protein 51 by Conformational Control. J. Med. Chem. 2013, 56 (10), 3922–3935. https://doi.org/10.1021/jm400087k.
- Wavefunction and Reactivity Study of Benzo[a]Pyrene Diol Epoxide and Its Enantiomeric Forms. Struct. Chem. 2014, 25 (5), 1521–1533. https://doi.org/10.1007/s11224-014-0430-6.
分子芳香性和离域性分析服务通过计算分子的π电子分布和离域能量,帮助客户理解分子的芳香性、电子离域程度及共轭体系的稳定性,进一步分析其对化学稳定性和反应活性的影响。
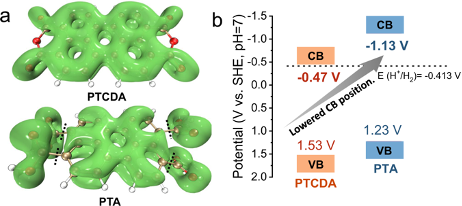
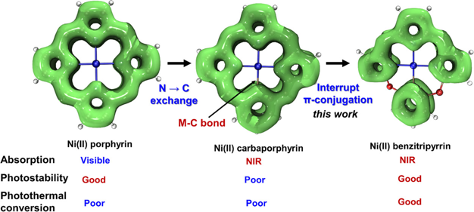
半导体材料的共轭性分析 【1】 离域区域分析【2】
- Perylenetetracarboxylic Acid Nanosheets with Internal Electric Fields and Anisotropic Charge Migration for Photocatalytic Hydrogen Evolution. Nat. Commun. 2022, 13 (1), 2067. https://doi.org/10.1038/s41467-022-29826-z.
- Nonaromatic Organonickel(II) Phototheranostics. J. Am. Chem. Soc. 2022, 144 (16), 7346–7356. https://doi.org/10.1021/jacs.2c00710.
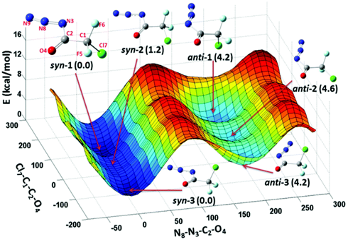
本研究实现铑(III)催化的环己二烯酮连接的烯烃(1,6-二烯)与B2pin2之间的不对称硼基化环化,生成了具有三个或四个相邻立体中心的光学纯顺式氢化苯并呋喃骨架。DFT计算的反应机理揭示了一个涉及双硼基铑(III)中间体的Rh(I)–Rh(III)催化循环。该反应通过烯烃插入、共轭加成和随后的还原消除步骤进行,其中,环化共轭加成是决定区域选择性的关键步骤。主要结果如下【1】
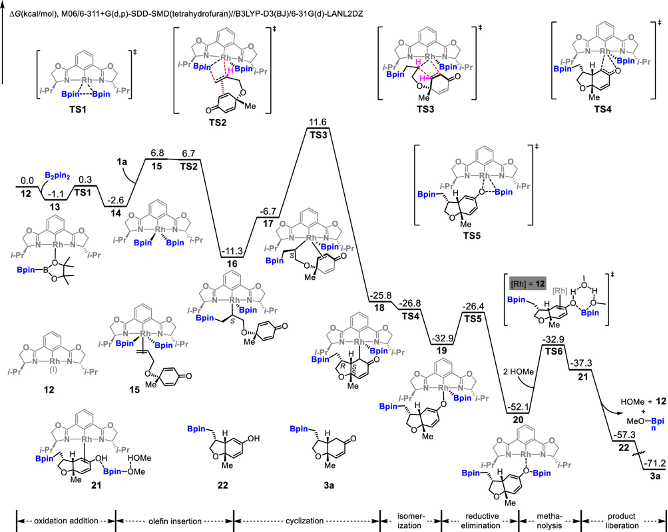
完整反应路径能量折线图
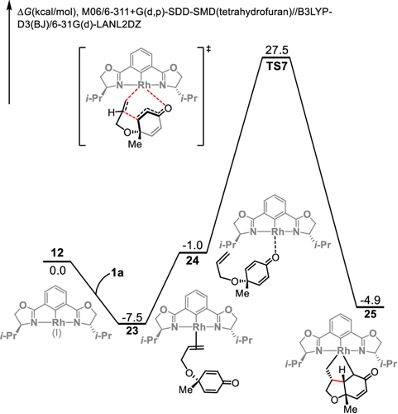
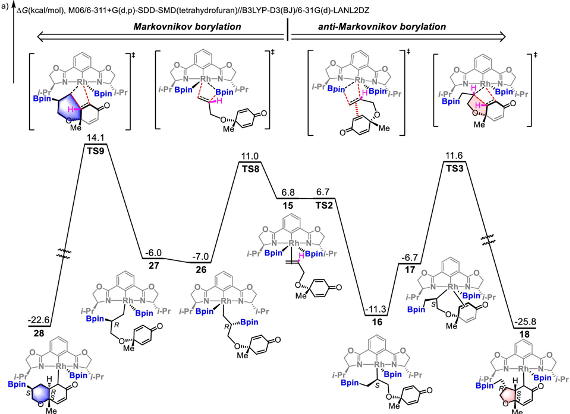
可能的反应路径B 马氏/反马氏产物竞争反应机理
- Rhodium(III)-Catalyzed Asymmetric Borylative Cyclization of Cyclohexadienone-Containing 1,6-Dienes: An Experimental and DFT Study. J. Am. Chem. Soc. 2019, 141 (32), 12770–12779. https://doi.org/10.1021/jacs.9b05583.
交付内容
交付内容